atomistica.online
MOLECULAR MODELING COURSE
Introduction to Property Calculations and Post-Processing
What are Property Calculations and Post-Processing of .out Files?
Within molecular modeling, understanding the energy and structure of molecules is foundational. But to dive deeper, property calculations enable the prediction of a wide variety of molecular properties. These range from electric and magnetic properties to complex spectral data. Additionally, the raw results of these calculations, often encapsulated in .out files, can be processed further with specialized tools. This post-processing unveils and visualizes a plethora of additional molecular properties, most notably the Molecular Electrostatic Potential (MEP) and Average Localized Ionization Energy (ALIE) surfaces.
Why are They Important?
Predictive Power: Property calculations offer a glimpse into potential experimental outcomes, thereby guiding experimental design.
Deep Molecular Insights: Through properties like polarizability or MEP surfaces, one can understand molecular interactions and behaviors in greater depth.
Guide to Design: By predicting molecular properties, researchers can tailor materials for specific applications, be it drug development or novel catalysts.
Spotlight on Reactivity: Advanced insights, such as those from ALIE surfaces, can pinpoint potential reactive sites in molecules.
Validation & Refinement: By comparing calculated properties to experimental data, the chosen modeling methods can be validated or refined for accuracy.
How Does it Work?
Starting with Optimized Geometry: An optimized molecular geometry often serves as the foundational step for both property calculations and post-processing.
Choice of Method: The desired property often dictates the quantum mechanical method or computational strategy. For instance, electronic spectra might rely on TD-DFT, while NMR shifts could employ GIAO methods.
Calculation & Data Extraction: Property evaluations are carried out based on the given molecular configuration. Concurrently, .out files can be mined for pertinent data using specialized software.
Advanced Data Processing: This extracted data is then processed further. For example, MEP surfaces might be sculpted based on electron densities and atomic charges.
Visualization: Modern tools facilitate the creation of vivid 3D molecular renderings, showcasing properties like MEP or ALIE for deeper insights.
Points to Remember:
It’s crucial to ensure that .out files contain comprehensive data for post-processing. Some calculations might require tailored settings to capture all essential details.
Selection of software tools should consider compatibility with molecular modeling software and specific research objectives.
Visualization serves not just for understanding but is invaluable for presentations and publications.
The fidelity of property calculations is tethered to the chosen method and basis set.
External factors, like solvents, can significantly influence property predictions.
High computational demands might arise, especially for complex systems or when using advanced methods.
Practical part 1 - Calculations of electron affinity
Overview
In this practical exercise, we will use ultra fast GFN2-xTB method implemented in the xTB program to calculate some properties of the [6,6]-Phenyl-C61-butyric acid methyl ester (PCBM) molecule. Later, we will use results of DFT calculations on PCBM obtained with ORCA modeling program to produce molecular electrostatic potential surface (MEP), and to calculate standard quantum-molecular descriptors. The purpose of this exercise is to learn that some properties can be calculated and obtained directly from calculation, while some properties can be obtained using the external tools, such as Avogadro program or tools of atomistica.online. First, lets deal with PCBM molecule. You can download its structure using the button bellow.
To download, right click and click “Save Link As”. When downloading .out files, WordPress changes the extension from .out to .txt. Revert it manually to .out.
3D structure of PCBM:
The provided structure of PCBM is already optimized, so there is no need to perform optimizations in the calculations that are going to be done. Before we proceed, a few words about this interesting derivative of fullerene. It comprises of 88 atoms, so dealing with this molecule using DFT calculations would be time-consuming. Luckily, xTB program and therein implemented GFN2-xTB method allow quick calculation of some properties. This molecule is of importance for solar cells.
PCBM is commonly used as the electron acceptor material in bulk heterojunction organic solar cells. Its unique spherical structure and high electron affinity make it an ideal candidate for efficiently accepting and transporting electrons generated from absorbed photons in these solar cells. PCBM’s combination of good electron mobility, compatibility with various conjugated polymers, and ease of processing have contributed to its widespread use in organic photovoltaic devices, helping advance renewable energy technologies. Great thing about atomistic calculations is the fact that we can use them to evaluate electron affinity. With xTB program and GFN2-xTB method, you can evaluate this important parameter within seconds for a large molecule such as PCBM. Of course, one should be aware of the limitations of the GFN2-xTB method, but it is a good starting point, especially for teaching purposes. So let us first calculate electron affinity of PCBM using Online xTB calculator.
Procedure for calculating electron affinity
Step 1 – Download molecular structure of optimized PCBM
Step 2 – Open “Online xtb calculator” to perform calculations of electron affinity
Step 3 – Load the structure of optimized PCBM
Step 4 – Select task: Energy
Step 5 – Select method: GFN1-xTB (we use this method because this method is adjusted for calculations of electron affinity)
Step 6 – Click on button “SHOW ADVANCED SETUP”
Step 7 – Tick “Properties” check box
Step 8 – Tick “Electron Affinity” check box
Step 9 – Press “RUN XTB” button. This will now make a single point energy calculation with calculation of electron affinity
Step 10 – Inspect the produced output file to find a value of electron affinity. Search for “delta SCC EA (eV)”. What is the value of electron affinity.
Step 11 – Compare the calculated electron affinity value with the experimental value. A value of electron affinity experimentally determined in J. Phys. Chem. C 2013, 117, 29, 14958–14964 was 2.63(1) eV.
Homework
Do the calculation of ionization potential for pentacene molecule. You can download the optimized structure of pentacene by using the button bellow.Pentacene is one more well known molecule in organic electronics, typically used as an electron donor. This means it should have representative values of ionization potential. So, for the homework, calculate
Pentacene is one more well known molecule in organic electronics, typically used as an electron donor. This means it should have representative values of ionization potential. So, for the homework, calculate its ionization potential using the procedure given above. The only differences will be in steps 8 and 11. In step 8, instead of “Electron Affinity”, tick “Ionization potential”, while in Step 11, instead of searching for “delta SCC EA (eV)”, search for “delta SCC IP (eV)”. Again, please compare the calculated and experimentally determined values. You can fine experimental values of ionization potential for pentacene at the NIST webpage dedicated to pentacene.
Practical part 2 - Reactivity descriptors with xTB
Overview
In this part you will learn how to calculate the so-called global reactivity descriptors, that are frequently employed in scientific papers. Once you perform single-point energy calculation, you obtain the information about HOMO and LUMO Energies. The HOMO represents the highest energy orbital with electrons, while the LUMO represents the lowest energy orbital without electrons. Their energy values reflect the ease of donating (HOMO) or accepting (LUMO) electrons in a chemical reaction. Using information about HOMO and LUMO energies, one can use a set of simple equations to calculate quantities that reflect the global reactivity of a molecular system. In these regards, we will mention global descriptors, such as hardness (η), softness (S), chemical potential (μ) and electrophilicity.
$$ \eta \approx \frac{1}{2} \cdot (E_{LUMO}-E_{HOMO})$$
$$ S \approx \frac{1}{\eta} $$
$$ \mu \approx \frac{1}{2} \cdot (E_{HOMO} + E_{LUMO}) $$
$$ \omega \approx \frac{\mu^2}{2\cdot\eta} $$
While η describes molecule’s stability (higher values mean higher stability), S describes how molecule responds to changes in electron density (softer system is more responsive to electron density variations, meaning it can more easily adapt to changes in electron density), μ describes molecule’s ability to attract electrons (as μ becomes more negative, it is more difficult for a molecule to lose an electron, and becomes easier to gain one), while ω indicates the electrophilicity (higher values mean higher electrophilicity).
Task
So, let’s calculate and compare quantum molecular descriptors of PCBM and pentacene, using “Online xTB calculator”. Taking into account that by now you collected some experience with calculations 🙂 there will be no detailed procedure. Perform single point energy calculations on optimized structures of PCBM and pentacene using GFN2-xTB method, and search for values of HOMO and LUMO in the output file (they are located close to the end of the file). Tabulate the results and try to make some conclusions when comparing these two molecules. Which molecule seems to be more stable?
Practical part 3 - Reactivity descriptors with ORCA calculation and output analyzer of atomistica.online
Overview
It is evidently not difficult at all to obtain information about global reactivity descriptors. It is enough dig into the output file and find HOMO and LUMO energy values, and with a few simple calculations you get parameters that are frequently analyzed in scientific literature. One thing that should be emphasized regarding previous calculations is that you should be aware that we used semiempirical calculations. While these calculations provide wonderful results, you may want to apply higher level calculations to obtain more accurate information about electron density and energies of molecular orbitals. For these purposes, DFT calculations are applied most frequently, but it is important to mention that highly accurate wavefunction methods are available for these purposes (but they might be computationally expensive). In this part, you will use output files of single point energy calculations via DFT method (B3LYP functional and 6-31G(d,p) basis set) for PCBM and pentacene molecules. Again, you will use the already obtained output files produced by calculations in ORCA, that can be downloaded bellow.
To download, right-click and click “Save Link As”. When downloading .out files, WordPress changes the extension from .out to .txt. Revert it manually to .out.
Procedure
Step 1 – Download output files of single-point energy calculations for PCBM and pentacene
Step 2 – From the Homepage of atomistica.online, open “Online Output File Analysis”
Step 3 – Load the output file (for example, load the pcbm_spe.out first)
Step 4 – Press “GET DATA” button, and you will get the values of all reactivity descriptors.
Yes, it’s that easy 🙂
Do the same for pentacene_spe.out file and get reactivity descriptors. Compare the values and comment the obtained results. If you feel motivated 🙂 tabulate the results and compare with the results obtained via GFN2-xTB method.
Practical part 4 - Visualization of Orbitals and MEP
Overview
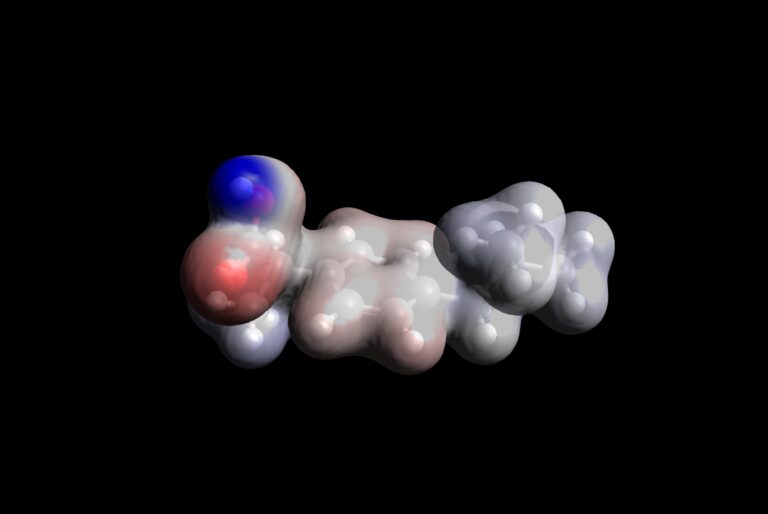
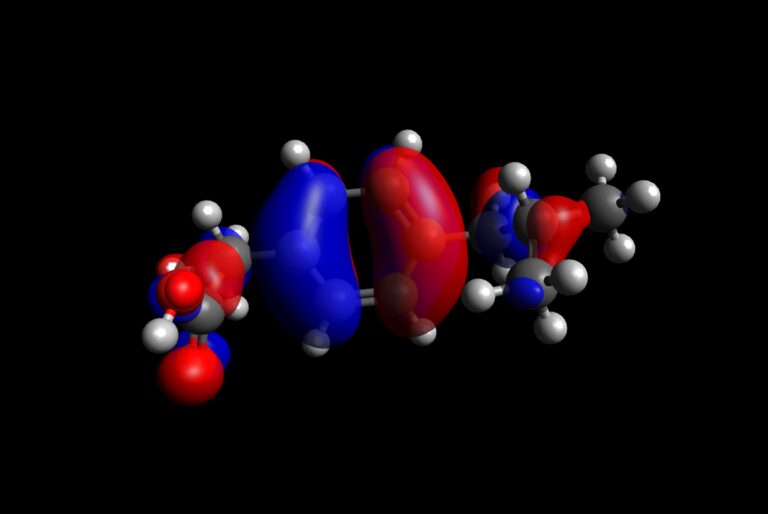
Scientific publications involving molecular modeling very frequently contain visualization of HOMO and LUMO orbitals, and molecular electrostatic potential (MEP) surfaces. As mentioned before, HOMO shows the places within molecule with abundance of electrons, while LUMO shows places within molecule where electrons can be hosted. This is extremely important to understand reactivity of molecule. Additionally, from the aspect of electrostatic interactions, it is also extremely useful to know where the MEP descriptor has lowest and highest values. Now, for these purposes, we will use the output file of single-point energy calculation on ibuprofen, because it is a smaller molecule than PCBM, and more interesting (in terms of reactivity) than pentacene 🙂 . You can download it using the button bellow.
To download, right-click and click “Save Link As”. When downloading .out files, WordPress changes the extension from .out to .txt. Revert it manually to .out.
Procedure to visualize frontier molecular orbitals
- Open Avogadro
- Load output file obtained by DFT calculations on ibuprofen molecule
- On the right, you should automatically get the possibility to select which molecular orbitals to plot.
- Scroll down to HOMO and LUMO orbitals and select them. Avogadro should immediately plot them.
- Comment the results in terms of charge transfer
Procedure to visualize MEP
- Open Avogadro
- Load output file obtained by DFT calculations on ibuprofen molecule
- Go to Extensions\Create Surfaces\ that will open the “Create Surfaces” window
- Select the following parameters:
Surface Type: Electron Density
Color By: Electrostatic Potential
Resolution: Medium
Iso Value: 0.01
In Display Type: Surfaces - Now press “Calculate” button. The calculation will take 5-10 seconds on a standard computer and you will get MEP surface
- Red color corresponds to the lowest negative values of the MEP, while blue color corresponds to the highest positive values of MEP
- Conclude which atoms of ibuprofen seem to most reactive from the aspect of electrostatic interactions.