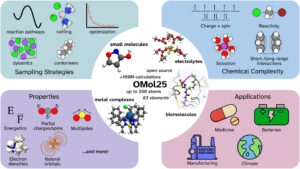
ATOMISTICA Scientific and commercialSoftware development Choose atomistica solutions Scientific Research Solutions Online and desktop applications for atomistic calculations completely FREE for non commercial academic purposes Professional & Industry Services Atomistica is a registered business entity providing a wide range of software solutions Education & Training Educational resources of atomistica related to computational modeling with atomistic calculations Technology & Platforms Blog and news portal dedicated to diverse topics in science and technology Read the latest articles CPUs for Quantum Mechanical Calculations on Budget (in 2025) Read More Mini PCs and NUCs Read More Synopsis Acquires Ansys Read More OMol25 and UMA Read More Load More